B. A.
Maiyarchuk1* and I. B. Rogozin23
1 Institute of Biological Problems
of the North, Far-East Branch of the Russian Academy of Sciences, Portovaya
str. 18, 68500 Magadan, Russia
2Institute of Cytology and Genetics,
Siberian Branch of the Russian Academy of Sciences,
630090 Novosibirsk, Russia 3 National Center for Biotechnology
Information, NLM, National Institutes of Health, Bethesda MD
20894, USA
Summary
To study spontaneous base substitutions in human mitochondrial DNA (mtDNA), we reconstructed the mutation spectra of the hypervariable segments I and II (HVS I and II) using published data on polymorphisms from various human populations. Classification analysis revealed numerous mutation hotspots in HVS I and II mutation spectra. Statistical analysis suggested that strand dislocation mutagenesis, operating in monotonous runs ofnucleotides, plays an important role in generating base substitutions in the mtDNA control region. The frequency of mutations compatible with the primer strand dislocation in the HVS I region was almost twice as high as that for template strand dislocation. Frequencies of mutations compatible with the primer and template strand dislocation models are almost equal in the HVS II region. Further analysis of strand dislocation models suggested that an excess of pyrimidine transitions in mutation spectra, reconstructed on the basis of the L-strand sequence, is caused by an excess of both L-strand pyrimidine transitions and H-strand purine transitions. In general, no significant bias toward parent H-strand-specific dislocation mutagenesis was found in the HVS I and II regions.
Keywords: mitochondrial DNA, spontaneous substitution, dislocation mutagenesis, context, mutation hotspot
Introduction
The origin of mitochondrial DNA (mtDNA) mutations, both inherited and somatic, is one of the most important questions in evolutionary and population mitochondrial genetics, as well as in molecular medicine, since mtDNA mutations are related to a variety of human degenerative diseases and cancer (Wallace, 1999; Copeland et al. 2002). The human mitochondrial genome is a small (16569 base pairs) double-stranded circular molecule and is strictly maternally inherited (Giles et al. 1980; Anderson et al. 1981). Human mtDNA evolves rapidly, at a rate 5-10 times higher than single-copy nuclear genes (Brown, 1980).
Corresponding author: Dr. Boris A. Malyarchuk, Genetics Laboratory, Institute of Biological Problems of
the North, Portovaya str., 18, 685000 Magadan, Russia. Fax/Phone: 7 41322 34463. E-mail: malyar@ibpn.kolyma.ru
Multiple copies of mtDNA are present in cells and within each mitochondrion. The copy number of mtDNA is typically 100-10000 copies/cell, depending on cell type (Wallace, 1999). Several possible factors that may cause the high mutation rate in mtDNA including inefficient DNA repair systems, a lack of DNA protective proteins, and continuous exposure to the muta-genic effects of reactive oxygen species (ROS) generated by oxidative phosphorylation (Richter et al. 1988; Copeland et al. 2002). However, recent studies have shown that mammalian mitochondria are well equipped to conduct base-excision repair (Bogenhagen, 1999; Dianov et al. 2001) and probably mismatch repair (Mason et al. 2003). A lack of histones covering the entire mtDNA molecule is probably equilibrated by the presence of the multifunctional protein TFAM (mtTFA, mitochondrial transcription factor A), which may cover the entire mtDNA, but preferentially binds to an active promoter region and cruciform DNA
structures (Kang & Hamasaki, 2002). Ribonucleotides covering the parent H-strand, which remains single-stranded for a long time during mtDNA replication and consequently might be damaged at a higher rate than the L-strand (Reyes et al. 1998), may also carry out a protective function, because it was recently found that replication intermediates contain large regions of RNA:DNA hybrid as a result of the incorporation of ribonucleotides on the lagging L-strand during mtDNA replication (Yang et al. 2002). Another factor explaining the high mutation rate in mtDNA is spontaneous errors arising during DNA replication. Although the fidelity of mtDNA polymerase is very high, it has been shown to cause frameshift errors in homopolymeric runs (Longley et al. 2001). It has further been that defects of nuclear genes responsible for mtDNA replication and maintenance cause the accumulation of mtDNA mutations (Copeland et al. 2003).
Most mtDNA variability studies have been examining sequence variation of the fast-evolving major non-coding (or control) region, which spans 1122 bases between the tRNA genes for proline (tRNAPro) and phenylalanine (tRNAPhe) (Anderson et al. 1981). This region is highly polymorphic, and the majority of mutations are concentrated in two hypervariable segments, HVS I (positions 16024-16365) and HVS II (positions 73-340). Results of phylogenetic studies have suggested a complex pattern of mtDNA control region evolution. It was found that base composition in the HVS I and II regions is not uniform, transitions occur with higher frequencies compared to transversions, the number of pyrimidine transitions in the L-strand exceeds the number ofpurine transitions, and substitution rates vary among nucleotide positions (Hasegawa et al. 1993; Wakeley, 1993; Excoffier & Yang, 1999; Meyer et al. 1999; Heyer et al. 2001; Pesole & Saccone, 2001; Malyarchuk et al. 2002b). Strand slippage in repetitive sequences may result in base substitutions by the transient misalignment dislocation mechanism. This model suggests that transient strand slippage in a monotonous run of nucleotides in the primer or template strand is followed by incorporation of the next correct nucleotide (Kunkel, 1985). Our analysis of phylogenetically reconstructed mutation spectra (distributions of mutations along analyzed sequences) of the mtDNA HVS I and II regions has suggested that dislocation mutagenesis plays an important role in generating base substitutions in mtDNA (Malyarchuk et al. 2002b). Additionally, it was found that next-nucleotide effects and dislocation mutagenesis may contribute to the formation of mtDNA mutations in patients with alterations of nucleoside metabolism (Nishigaki et al. 2003). Therefore the origin ofmtDNA mutation hotspots may, to a great extent, depend on the contextual properties ofthe mtDNA. To study spontaneous base substitutions in human mtDNA, we reconstructed mutation spectra for the HVS I and II regions using published data on polymorphisms in various human populations and analyzed them by means of the dislocation mutagenesis model (Kunkel, 1985; Kunkel & Soni, 1988).
Materials and Methods
Reconstruction of Mutation Spectra
To determine mutations in the mtDNA control region, we have analysed different phylogenetic haplogroups of mtDNA revealed by means ofmedian network analysis (Bandelt et al. 1995; http://fluxus-engineering.com for Network 3.1 program). We only used published population data comprising both the HVS I and/or HVS II nucleotide sequences and additional RFLP or coding-region information for each haplotype. Coding region variation was used to assign control region sequences to the phylogenetic haplogroups and place them in the reconstructed mtDNA phylogenetic tree (Macaulay et al. 1999; Maca-Meyer et al. 2001; Finnila et al. 2001; Herrnstadt et al. 2002; Kivisild et al. 2002; Salas et al. 2002; Yao et al. 2002). Using the human mtDNA nomenclature (Richards et al. 1998; Macaulay et al. 1999), each major clade (or mtDNA haplogroup) and nested subclade (or subhaplogroup) ofthe mtDNA tree was denoted with the corresponding Roman numerals.
The HVS I data set comprised 7482 sequences (between positions 16092-16365) belonging to 90 continental-specific mtDNA haplogroups and subgroups. This data set includes 3834 HVS I sequences from 28 West Eurasian haplogroups and subgroups: H, HV*, pre-V pre-HV R*, T1, T* ,J*, J1a, J1b, J2, K, U*, U1, U2, U3, U4, U5, U7, U8a, U8b, N1a, N1b, N1c, N* , I, W, X (according to data of Richards et al. 2000); 801 sequences from 34 East Eurasian haplogroups and subgroups: C, Z, M8a, D* (including D4), D5, G2, G3, G4, E, M* ,M7*, M7b, M7c, M9, M10, A, N9a, N2, N*, Y, R9a, R*,F*, F1a, F1b, F1c, F2, B*, B4* , B4a, B4b, B5* , B5a, B5b (according to data from Derbeneva et al. 2002a, 2002b; Kivisild et al. 2002; Yao et al. 2002; Derenko et al. 2003); and 2847 sequences from 28 African haplogroups and subgroups: L1a1, L1a2, L1b, L1c*, L1c1, L1c2, L1c3, L1d, L1e, L2a* , L2a1a, L2a1b, L2b, L2c, L2d1, L2d2, L3b1, L3b2 (including L3b* ), L3d, L3e1, L3e2, L3e3, L3e4, L3f* , L3f1, L3g, U6, M1 (according to the data from Salas et al. 2002). Note that haplogroups U6 and M1 were included in the African-specific data set, because haplogroup U6 has predominantly been found in North Africans (Macaulay et al. 1999) and haplogroup M1 may have originated in East Africa (Quintana-Murci et al. 1999).
The HVS II data set was represented by 1703 individual sequences (between positions 72-297) belonging to 71 mtDNA haplogroups and subgroups. This data set includes 1002 HVS II sequences from 27 West Eurasian haplogroups and subgroups: H, HV* , pre-V, pre-HV R*, T1, T* ,J*, J1a, J1b, J2, K, U*, U1, U2, U3, U4, U5, U7, U8a, U8b, N1a, N1b, N1c, I, W, X (according to data ofHoffman et al. 1997; Finnila et al. 2001; Derbeneva et al. 2002a; 2002b; Malyarchuk et al. 2002a; Derenko et al. 2003); 496 sequences from 33 East Eurasian haplogroups and subgroups: C, Z, M8a, D* (including D4), D5, G2, G3, G4, E, M*,M7*, M7b, M7c, M9, M10, A, N9a, N*, Y, R9a, R* ,F*, F1a, F1b, F1c, F2, B* ,B4*, B4a, B4b, B5*, B5a, B5b (according to data ofFinnila et al. 2001; Derbeneva et al. 2002a; 2002b; Kivisild et al. 2002; Malyarchuk et al. 2002a; Yao et al. 2002; Derenko et al. 2003); and 205 sequences from 11 African haplogroups and subgroups: L1a, L1b, L1c, L2a, L2b, L2c, L2d, L3*, L3e1, L3e2, L3e3 (according to data from Alves-Silva et al. 2000; Chen et al. 2000; Bandelt et al. 2001; Maca-Meyer et al. 2001; Torroni et al. 2001).
Mutations were inferred from the mtDNA median networks constructed, following the guidelines of Bandelt et al. (2000), and verified by comparison with the published networks. Parallel mutations in mtDNA were inferred by revealing variable positions in which identical mutations arose independently in different mitochondrial haplogroups or subgroups, as described by Macaulay et al. (1999), Finnila et al. (2001), Malyarchuk & Derenko (2001) and Herrnstadt et al. (2002). In all cases we have studied mutation spectra represented by nucleotide substitutions; therefore, nucleotide positions that showed point insertions or deletions were excluded from the analysis. All mutations are described using the L-strand orientation and numbered according to the mtDNA Cambridge Reference Sequence (CRS; Anderson et al. 1981; Andrews et al. 1999).
Hotspot Prediction
The general principle of mutation hotspot prediction in this study was based on a threshold (Sh) value for the number of mutations in a mutable site. All sites with the number of mutations greater than or equal to Sh were defined as hotspots. The threshold value and the resulting hotspot sites were defined for each mutation spectrum separately, using the CLUSTERM program (www.itb.cnr.it/webmutation/; Glazko et al. 1998). Briefly, this program decomposes a mutation spectrum into several homogeneous classes ofsites, with each class approximated by a Poisson distribution. Variations in mutation frequencies among sites of the same class are random by definition (mutation probability is the same for all sites within a class), but differences between classes are statistically significant. A class (or classes) with the highest mutation frequency is(are) called a hotspot class(es). Each site has a probability P(C) assigned to a class C. Sites with P(Chotspot) > 0.95 in hotspot class Chotspot are defined as hotspot sites. This approach ensures that the assignment is statistically significant and robust (Glazko et al. 1998; see Rogozin et al. 2001 for details).
Statistical Analysis of Dislocation Model
Wrandom is the same or greater than W. Low probability values (PW<Wrandom ≤0.05) indicate a good correspondence between the mutation data and the analyzed dislocation model.
Substitution Frequencies and Strand Asymmetry
Reconstructed mutation spectra in the HVS I and II regions are shown in Figures 1 and 2, respectively. The first reconstructed spectrum includes 2212 substitutions in 202 variable nucleotide positions, while the second


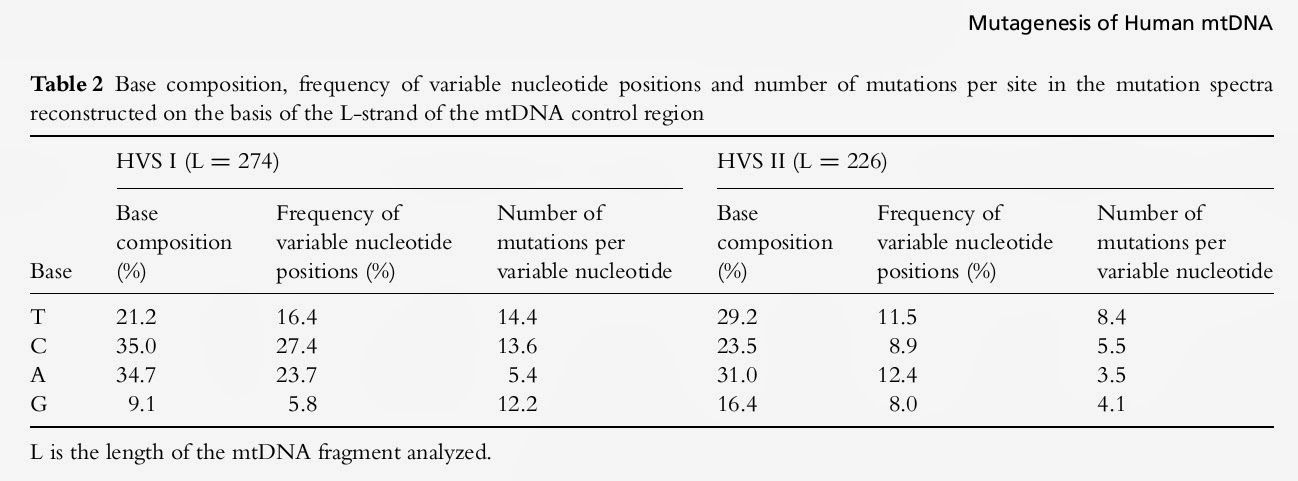
There is a good correspondence between base composition and the frequency of variable nucleotide positions: the higher the content of a certain nucleotide, the higher the frequency of changed nucleotides observed (Table 2). The correlation between base content and frequency of variable positions was highly significant (P < 0.01) for both the HVS I and HVS II data sets (r = 0.99 and 0.96, respectively). Nevertheless, the average number ofmutations (both transitions and transversions) per site varies considerably, with the lowest values observed for adenine, despite the high content of this base in the L-strand of HVS I and HVS II sequences. This conclusion is also valid when one compares the distributions of the variable nucleotide positions with the number of independent mutations occurring there (Table 1 and 2).
Mutation Hotspots
Analysis of the mutation spectrum in the HVS II region using CLUSTERM revealed four classes of sites. The first class includes obvious "cold" sites, with the number of substitutions varying from 0 to 3; the second class includes sites with the number of mutations from 0 to 10; the third class includes sites with the number ofmutations from 7 to 20; the fourth class includes obvious hotspot sites where the number of mutations varied from 32 to 56. Differences between the observed and the expected distributions (a mixture of four Pois-son distributions) were statistically insignificant. Since the members of the third class (P(site in 3) > 0.95) were sites with more than 9 mutations, 10 was therefore chosen as the threshold value Sh for hotspot sites (these sites are underlined in Figure 2). Only 15 nu-cleotide positions out of 226 analyzed in the HVS II region were identified as hotspot sites using the classification analysis. Among them, the most frequent sites, which have undergone 32-56 changes, were 146, 150, 152 and 195. Sites 93, 151, 182, 185, 189, 194, 198, 199, 200, 204 and 207 were found to be very fast, with the means ofthe number ofchanges per site equal to 10-20.
Previous analysis ofthe HVS I mutation spectrum revealed four classes of sites, with 11 mutations used as the hotspot threshold (Malyarchuk et al. 2002b). The present study shows that 27% of sites in the HVS I region (74 out of 274 sites analyzed) should be classified as hotspots (these sites are underlined in Figure 1 and shown in Table 3). Among them, the fastest were sites 16093,16129,16172,16189,16291,16311 and 16362, which have experienced more than 40 changes per site.
+in+the+HVS+I+region.jpg)
It is noteworthy that almost all ofthese sites have already been identified as fast sites in phylogenetic and familial studies (Hasegawa et al. 1993; Wakeley, 1993; Excoffier & Yang, 1999; Meyer et al. 1999; Heyer et al. 2001; Pesole & Saccone, 2001; Bandelt et al. 2002). Thus, hotspot sites in the HVS I are now well defined, and results of the present and other analyses give a reliable description of variability in the HVS I region.
Dislocation Mutagenesis
It has been shown that transient misalignment dislocation mutagenesis operating in monotonous runs of nucleotides might play an important role in generating base substitutions in the mtDNA control region, and define its contextual properties (Malyarchuk et al. 2002b). This model suggests that transient strand slippage in a homonucleotide run in the primer or template strand is followed by incorporation of the next correct nucleotide (Kunkel, 1985). Present analysis of the HVS I and II spectra revealed that many base substitutions are consistent with the dislocation model. This model may explain the origin of 23.4% mutations (517 out of 2212) found in 34.7% of variable nucleotide positions in the HVS I (70 out of 202 sites) and of 49.4% mutations (247 out of 500) in 27.2% of variable positions (25 out of 92) in the HVS II region. For each hyper-variable region, transitions predominate over transver-sions, being found with a ratio of 448:69 (s/v = 6.49) and 247:0 in HVS I and II, respectively. As for hotspot distribution, the model of dislocation mutagenesis can explain the origin of 16% of hotspot sites (with the number of changes per site >10) in the HVS I region, whereas in the HVS II 40% ofhotspot sites (6 out of 15, including the most variable positions 146, 150, 152, and 195) may be caused by the strand dislocation model.
Recently, we have found that only the primer strand dislocation model has statistically significant support in both HVS spectra (Malyarchuk et al. 2002b). The present study partly supports this conclusion; however, important new details were revealed from analysis of the strand dislocation model itself. It is well established that mtDNA replication starts in the right domain ofthe control region (including the HVS II region) with the expansion ofthe displacement loop (D-loop), a stable triple-stranded structure (Clayton, 1982). During the D-loop formation, the parental H-strand is displaced by the nascent H-strand, remaining in a single-stranded state until L-strand DNA synthesis is initiated in the opposite direction to that of the H-strand. Thus, human mtDNA replicates by an unusual asynchronous transcription-primed mechanism that is initiated at the origin of H-strand replication (Shadel & Clayton, 1997). The mtDNA replication model suggests that the higher rate of transition between pyrimidines observed in the L-strand mutation spectra of the control region may be a result of both a high substitution rate of pyrimidine transitions on the L-strand, and purine transitions on the H-strand, or one may speculate that the observed high rate of pyrimidine transitions on the L-strand is a result of transitions between purines that occurred on the parental H-strand due to the probable higher mutability ofpurines on the single-stranded H-strand during mtDNA replication (Reyes et al. 1998; Tamura, 2000). However, this classical asymmetric replication model has recently been questioned by the finding that mammalian mtDNA replication is initially bidirectional, but after fork arrest near OriH, replication is restricted to one direction only (Bowmaker et al. 2003). The problem of determination of the strand of origin for some of the mutations observed in the HVS I and II mutation spectra can be solved using the strand dislocation model. Figure 3 demonstrates this for the Land H-strand mtDNA fragment (L: 5'-GATCCTA-3', H: 5'-TAGGATC-3') where monotonous dinucleotide CC/GG runs are located in the 5' and 3' direction with respect to T- and A-nucleotides on the L- and H-strands, respectively. Figure 3 shows that four different variants of primer and template strand dislocation at CC/GG runs during replication can lead to specific nucleotide substitutions at each position of the L-strand sequence 5'-TCCT-3'. As is seen, irrespective of which DNA strand is replicated, strand slippage in a homonucleotide run followed by base substitution occurs when homonucleotide runs are placed in a 3' direction. However, because ofthe complementary nature of DNA, we can observe a different location for homonucleotide runs relative to the variable nucleotide observed on the L-strand: homonucleotide runs are located in a 3' direction if strand dislocation originally occurred on the L-strand, and homonucleotide runs are located in a 5' direction if strand dislocation actually occurred on the H-strand (Figure 3). As a result, a simple rule predicting the mutation change, depending on which DNA strand is the strand of origin for the mutation and which strand (primer or template) was dislocated during replication, follows from the data obtained:
XYY — YYY (X→ Y, L-strand, primer strand) XYY — XXY(Y→ X, L-strand, template strand) YYX — YYY (X→Y, H-strand, primer strand) YYX — YXX(Y→ X, H-strand, template strand).
To determine the strands on which any observed transitions or transversions originally occurred as a result of strand dislocation mutagenesis, we have investigated the distribution of trinucleotide sequences, containing monotonous dinucleotide runs, on the L-strand of the mtDNA reference sequence. For transitions, the distribution of eight types of trinucleotides (TCC, CTT, AGG, GAA, TTC, CCT, AAG, GGA) was analyzed; for transversions, the distribution of 16 different



trinucleotide sequences (GTT, TGG, ATT, TAA, ACC, CAA, GCC, CGG, TTG, GGT, CCA, AAC, CCG, GGC, TTA, AAT) was examined. Taking into account the rules predicting mutations in accordance with the strand dislocation model, one may compare the distribution of variable positions and mutations observed in the HVS I and II sequence data sets with the expected distributions. Table 4 presents the observed and expected measurements for variable positions on each of the L and H strands for the HVS I data. The results suggest that more than half of the expected variable positions are actually observed on both mtDNA strands (53.3% on the L-strand and 61.9% on the H-strand). On the L-strand, pyrimidine transitions were most frequent, whereas on the H-strand purine transitions prevailed. It is important that there is strong statistical support for the primer strand dislocation model only for transitions occurring on the L-strand. Thus, for all 481 pyrimidine transitions observed on the L-strand (i.e., observed in the mutation spectra recorded relative to the L-strand), 256 (as pyrimidine transitions) originally occurred on the L-strand and 225 (as purine transitions) originated on the H-strand. Among 86 purine transitions observed on the L-strand, the overwhelming majority ofthem originally occurred on the H-strand as pyrimidine transitions (83 versus 3 purine transitions in the L-strand). For transver-sions, only 20.8% and 17.4% of the expected variable positions were found on the L and H strands, respectively. Among transversions, the most frequent on the L-strand were A-to-C and C-to-A substitutions (82% out of all transversions observed) and on the H-strand most common were G-to-T and T-to-G transversions (77.8%). Estimation of the primer/template ratios shows that only for the H-strand transversions is there a predominance of the template dislocation model over the primer model (primer/template ratio is 10/17); however, this excess is statistically insignificant (P = 0.12 by the binomial test).

Table 5 presents the observed and expected numbers of variable positions on the L and H strands for the HVS II sequence. These data suggest that less than half of the expected variable positions potentially involved in dislocation mutagenesis are actually observed on both mtDNA strands (41.7% on the L-strand and 23.8% on the H-strand). As in the case for the HVS I region, in HVS II most frequent were pyrimidine transitions on the L-strand and purine transitions on the H-strand. Strong statistical support for the primer strand dislocation model was found for transitions occurring on the H-strand, whereas template dislocation mutage-nesis operates three times more frequently on the L-strand. Among all 226 pyrimidine transitions observed in the L-strand mutation spectrum, 103 (as pyrimi-dine transitions) originally occurred on the L-strand and 123 (as purine transitions) on the H-strand. Among 28 purine transitions observed, the majority of them occurred on the L-strand (23 versus 5 transitions on the H-strand). We did not find the transversions compatible with the strand dislocation model in the HVS II region, although the expected number ofvariable positions potentially leading to transversions was high (116 and 110 positions expected on the L and H strands, respectively). In general, our results suggest that DNA strands dislocation during the replication appears to be an important mechanism for mutation in the mitochondrial genome. However, these mutational events may have a different origin depending on which DNA strand mutated. One of the interesting findings of the present study is the observation that there are some differences between the two mtDNA hypervariable regions, with respect to whether mutations occur on the primer or template strand during the replication. The frequency of mutations compatible with the primer strand dislocation in the HVS I region is almost twice as high as that for template strand dislocation (Table 4 and 5); no excess ofmutations compatible with primer strand dislocation was found in the HVS II. However, ifwe apply the dislocation mutagenesis model to mtDNA L and H strand replication, the primer dislocation mutagenesis, leading to pyrimidine transitions in the parental L-strand, may operate during H-strand replication primarily in the HVS I region, whereas in the HVS II the most pronounced mutation process is template strand dislocation mutagenesis, also generating pyrimidine transitions. It is noteworthy that during the L-strand replication only primer strand dislocation operates on the parental H-strand in the HVS I and II regions, leading to purine transitions.
Discussion
It is well known that the hypervariable segments ofthe mtDNA control region are characterized by extreme site-specific rate heterogeneity, with a high substitution rate at certain nucleotide positions called mutation hotspots (Hasegawa et al. 1993; Wakeley, 1993; Excoffier & Yang, 1999; Meyer et al. 1999; Pesole & Saccone, 2001; Bandelt et al. 2002; Malyarchuk et al. 2002b; Meyer & von Haeseler, 2003). In the present study, we have further extended this concept, analyzing the phylogenetically reconstructed mutation spectra of the HVS I and II regions. The results ofthis study clearly demonstrate that an excess ofpyrimidine transitions is a distinctive feature ofboth HVS I and II mutation spectra, and this is consistent with previous observations (Meyer et al. 1999; Tamura, 2000; Malyarchuk et al. 2002b).
However, despite the predominance ofpyrimidine transitions in both spectra, there is a good correspondence between the base composition and the frequencyofvari-able nucleotide positions. These results suggest that the variable positions in the HVS I and II regions may be free from selective constraints. Nevertheless, despite the high content of A-bases in the L-strand of the HVS I and HVS II regions, the average numbers of independent mutations per A have the lowest values among all bases analyzed, so the lower frequency of mutations at A-bases may be attributable to some (possibly, structural) constraints. It should be noted that according to our preliminary results for the mtDNA coding-region data (represented by about 800 sequences published in Finnila et al. 2001; Maca-Meyer et al. 2001; Herrnstadt et al. 2002), the pattern of nucleotide substitutions is different between the control region and the coding region. In the mtDNA coding region only 4%-10% of the sites appear to be variable, and there is no excess of pyrimidine over purine transitions. Meanwhile, despite the low content ofguanines in mtDNA as a whole, the highest numbers ofmutations per site are observed at Gs in the protein-coding and rRNA-coding genes, suggesting that mutational pressure at the nucleotide level might have an important role in generating base substitutions in the mtDNA coding regions. Thus, one of the interesting features of the mtDNA control region variability is a strong bias of nucleotide substitutions to pyrimidine transition changes. It has been suggested previously (Malyarchuk et al. 2002b), as well as in the present study, that DNA sequence context properties of the control region may influence the pattern ofsubstitutions seen in the HVS I and II mutation spectra. Previously, contextual analysis of hotspots has demonstrated a complex influence of neighbouring bases on mutagenesis in the HVS I region. Statistical analysis of these hotspots has revealed two hotspot motifs, CC and KTNCNK in the HVS I mutation spectrum. However, we have found that these motifs do not correlate with the distribution ofsubstitutions along HVS II. It seems that this might reflect some biological differences between mutation spectra in the HVS I and II regions. The most important finding of this and our previous study is that a statistically significant manifestation of dislocation mutagenesis for in vivo substitution spectra was found, indicating that such mutagenesis might be a general mechanism of substitutions in the human mtDNA control region. This model explains the origin of 23.4% and 49.4% of mutations observed in the HVS I and HVS II mutation spectra, respectively. Importantly, analysis ofthe strand dislocation model has allowed us to recognize the original DNA strands, L or H, on which the initial mutation events occurred. The data obtained have shown that the observed high rate of pyrimidine transitions in both HVS mutation spectra does not appear to be a result of the preferential purine transitions originating on the parental H-strand due, as previously speculated (Tamura, 2000), to the higher rate ofpurine transitions occurring on the single-stranded H-strand during mtDNA replication. We have found that both pyrimidine transitions on the L-strand and purine transitions on the H-strand give an approximately equal input to the excess of pyrimidine transitions observed in mutation spectra reconstructed on the basis ofthe L-strand sequence. It is possible also that these results are relevant to the model of mtDNA replication depicting an initially bidirectional replication, originating downstream of OriH (Bowmaker et al. 2003).
However, there should be other mechanisms ofmu-tation, since the strand dislocation model allows us to explain the origin ofonly source ofthe mutations seen in the HVS I and II mutation spectra. Among other possible mechanisms, depurination can explain the higher mutation rate of purines on the single-stranded H-strand, since the rate ofdepurination ofsingle-stranded DNA is four-times greater than that ofdouble-stranded DNA (Lindahl, 1993). In addition, the repair of abasic sites requires double-stranded DNA to use the complementary strand as the template (Pinz & Bogenhagen, 1998). Base loss, giving rise to apurinic/apyrimidinic sites, may be spontaneous or induced by oxidative stress DNA damage (Loeb & Preston, 1986). Deamination of cytosine to uracil and adenine to hypoxanthine is another possible mechanism ofmutation on the single-stranded H-strand (Reyes et al. 1998). This model suggests that one should expect more G-to-A and T-to-C transitions on the L-strand. The rates ofthese transitions are not so high in the HVS I (Table 1). Although the rate ofT-to-C mutations is higher in the HVS II region, the majority of them (73%) are compatible with the strand dislocation model. However, the compositional correlation with the duration ofsingle-stranded H strand during mtDNA replication is compatible with a gradient of deamination of cytosine to uracil and of adenine to hypoxanthine (Reyes et al. 1998). It has been found recently, by means ofuracil-N glycosylase treatment ofhu-man DNA extracted from archaeological sites, that postmortem damage ofancient mtDNA results mainly from the deamination ofcytosine to uracil, with a bias toward L-strand-specific C-to-T changes in the HVS I region (Gilbert et al. 2003). Note, however, that in vivo steady-state levels ofuracil (as well as hypoxanthine) in mtDNA have not yet been reported (Marcelino & Thilly, 1999). Moreover, relative to the nucleus, mitochondria contain a large excess of uracil-DNA glycosylase activity (~ 25% of the total cellular activity), scanning DNA for uracil residues (Bogenhagen, 1999). Thus, mitochondria may efficiently repair some DNA damage including uracil bases and abasic sites. Another type ofmtDNA damage may be due to ox-idative stress, since about 2% ofoxygen in mitochondria is converted to reactive oxygen species, which are able to cause endogenous oxidative damage to mtDNA, such as single- and double-strand DNA breaks, abasic sites and oxidized bases (Beckman & Ames, 1997). Of the oxidized bases, the most common and most important for inducing mutations is 8-oxoguanine that is characterized by a significantly higher incidence in mtDNA than in nuclear DNA (Richter et al. 1988). Nevertheless, there is strong evidence to suggest that mitochondria contain almost all components (e.g., 8-oxoguanine glycosylase, 8-oxoGTPase) of the system which responds to oxida-tive damage to guanosine, either as a free nucleotide or within DNA (Bogenhagen, 1999). In any case, as mtDNA is under strong oxidative stress, a significant excess ofC-to-A and G-to-T transversions (when G in a C:G pair is oxidized to 8-oxoG) and A-to-C and T-to-G transversions (when 8-oxoGTP is used as a nucleotide substrate during